
One validated platform, total compliance.
.png)




Traditional compliance is a bottleneck.



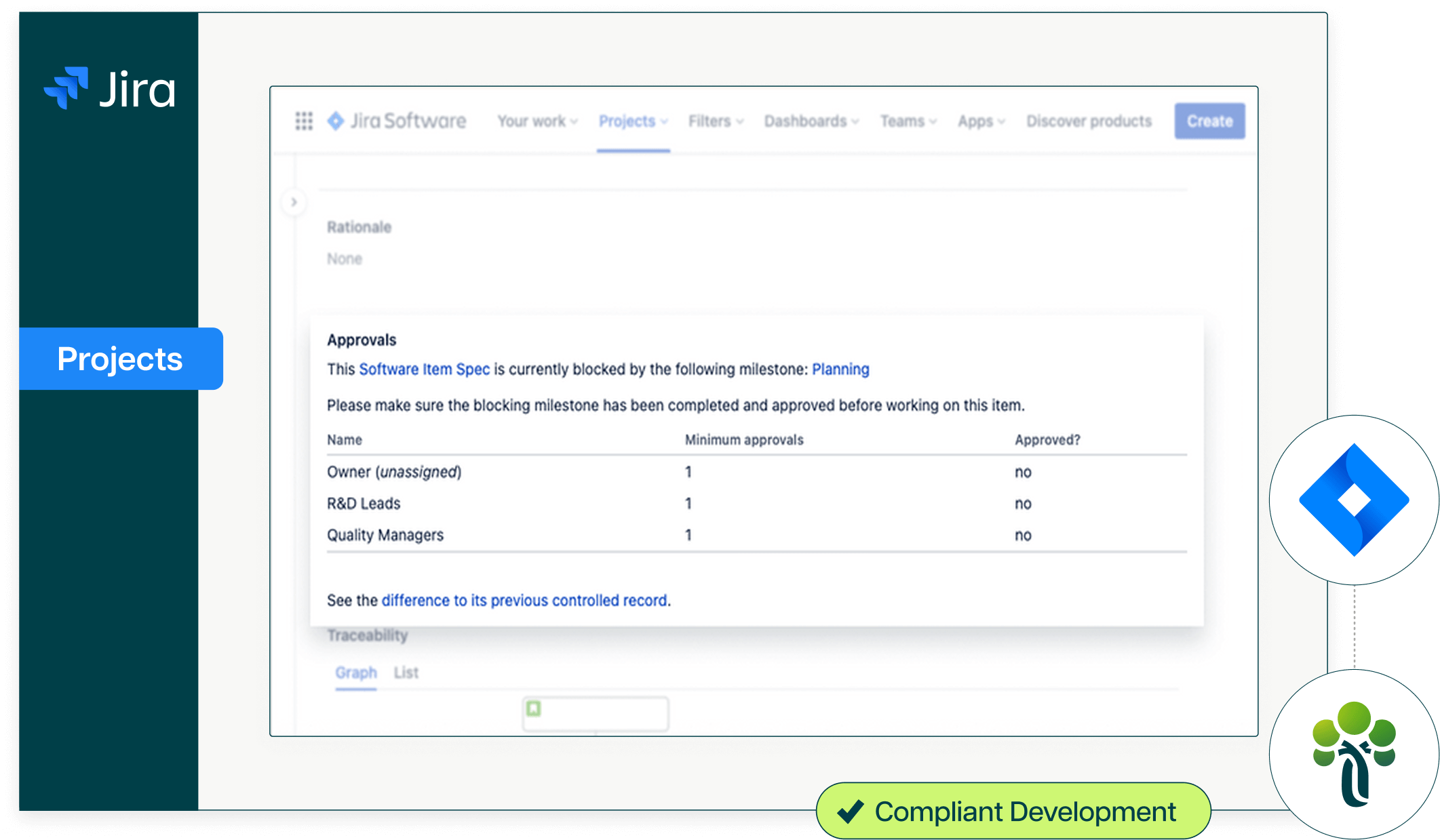
One platform that connects regulatory compliance to development.
Design history files generated automatically from development activity.
As your engineers write requirements in JAMA, push code to GitHub, and execute tests in TestRail, Ketryx generates your MDF deliverables, software requirements specifications, architecture documents, test protocols and reports, aligned to IEC 62304 and your submission format. Your MDF builds itself as the product progresses, not in a scramble before the FDA submission deadline.
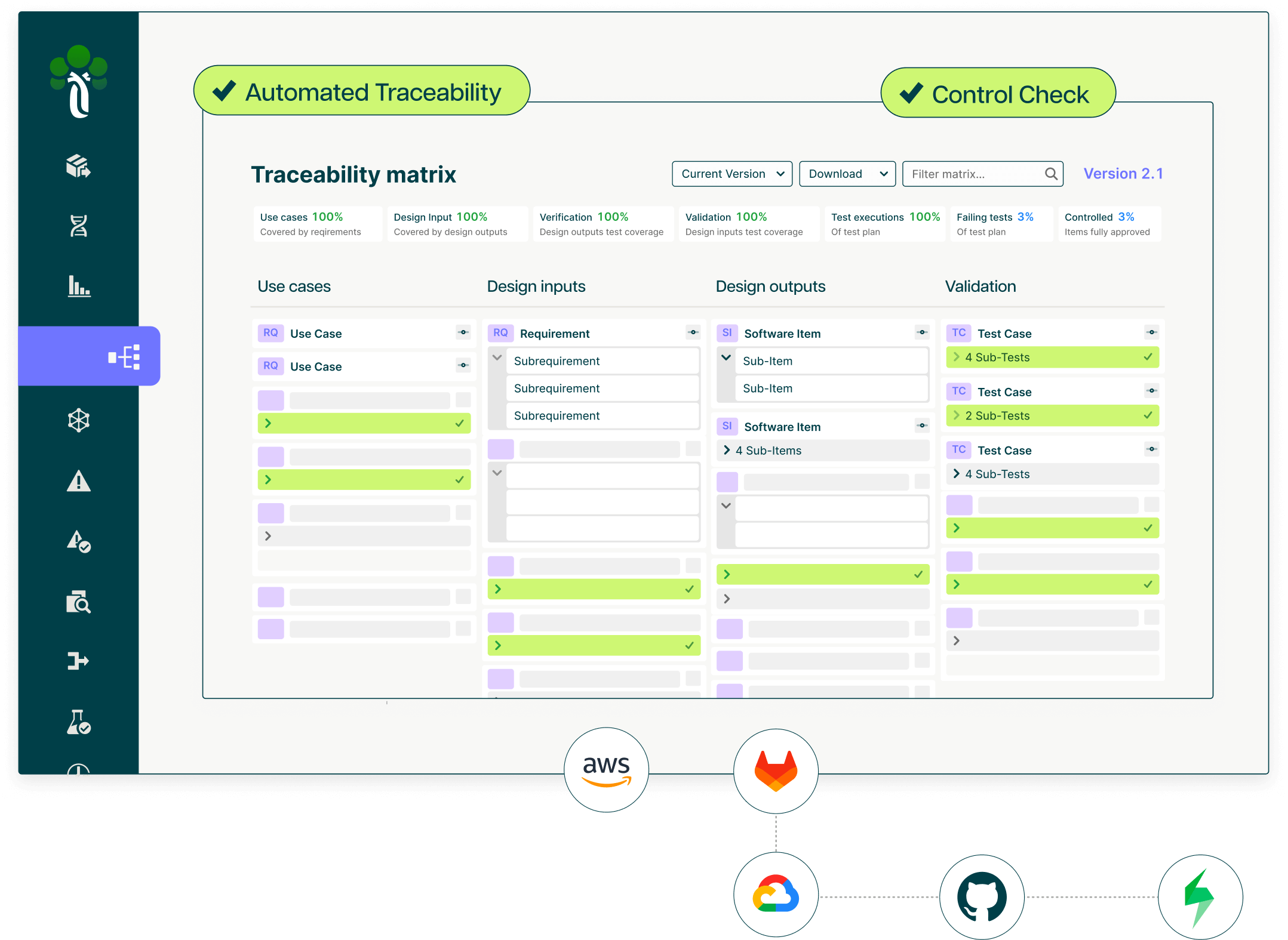
Live risk traceability from ISO 14971 hazard analysis to verified controls.
Ketryx connects your risk management file directly to design controls, verification evidence, and SOUP/OTS assessments. Every risk control is traced to its implementation and test result in real time. When a hazard's residual risk changes, because a test failed or a requirement shifted, your risk file updates automatically, not three months later.
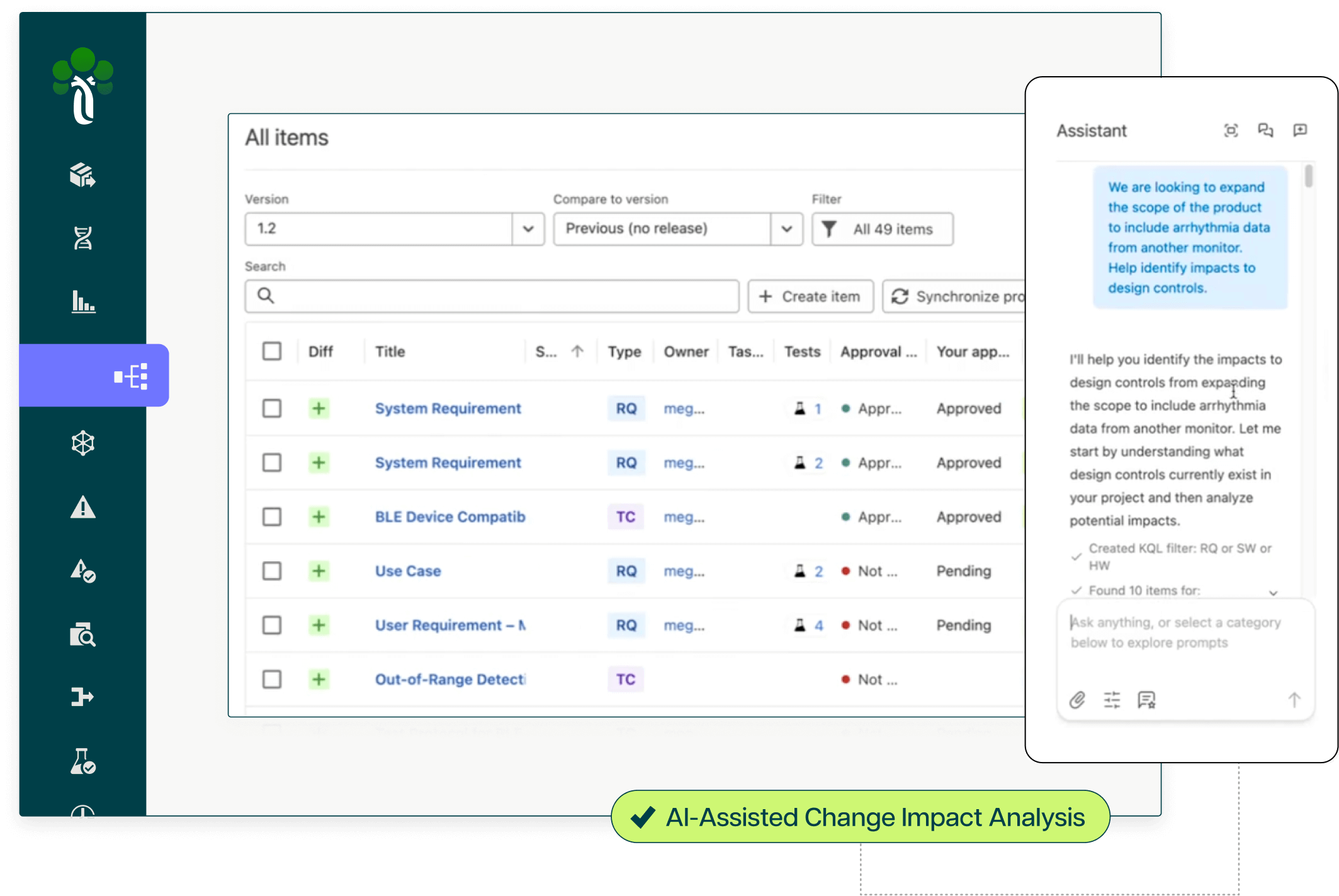
End-to-end traceability with AI agents to transform how your team scopes, reviews, and documents change impacts.
In minutes, the Ketryx Assistant provides a high-level overview of what a proposed change means in the context of your product, project, and team. No more opening dozens of documents to understand scope. Ketryx Agents analyze your MDF, QMS, and regulatory standards to provide specific recommendations. Experts review and take action with full traceability at every step. When a design input is modified, associated items are automatically flagged. The platform shows exactly what changed and what needs re-verification.
Trused by 4 of the Top 5 Medical
Device Leaders
Connects to the tools your team already uses.
Ketryx overlays your existing development and compliance tools — no rip-and-replace required. Your engineers stay in the tools they already use.

Built for IEC 62304, ISO 14971, ISO 13485, and 21 CFR Part 11
Frequently Asked Questions
Your engineers didn’t become engineers to spend their time assembling MDFs.
.png)