The FDA Just Published Free Advice for Building With AI. Here's What Your Team Needs to Know.
Table of Contents
Last week, the FDA told a pharmaceutical manufacturer that its use of AI in drug production was a violation. The company’s defense was, in essence, that the AI told them to do it this way. They didn’t know any better because they listened to the AI.
Most of the coverage I've seen reads this as a cautionary tale: AI is dangerous, slow down AI adoption, be careful.
"any output or recommendations from an AI agent must be reviewed and cleared by an authorized human representative of your firm’s [quality unit (QU)]" - FDA’s letter
I read it the opposite way. This is one of the most useful documents the FDA has published about AI in regulated industries. For the first time, they have told the industry precisely how compliant AI needs to look. That’s a design requirement, written in plain language and free for every team to read.
If you are building with AI inside a regulated lifecycle, this letter is a gift. You just have to read it as an architect.
The architecture lesson most people are missing
The company that received a citation was a small manufacturer, and some aspects of their practices (with or without AI) were clearly problematic. But the letter isn’t a story about bad actors or bad AI. This is a story about using AI without human oversight built into the architecture. These are completely different problems.
The FDA letter is a wake-up call for organizations using AI to produce content that affects product quality or informs regulatory decisions, whether that’s a batch record, requirement, risk control, or validation protocol. Every regulated team building with AI now has to answer the same question the FDA inspector was asking: who made the decision, and how can you prove it?
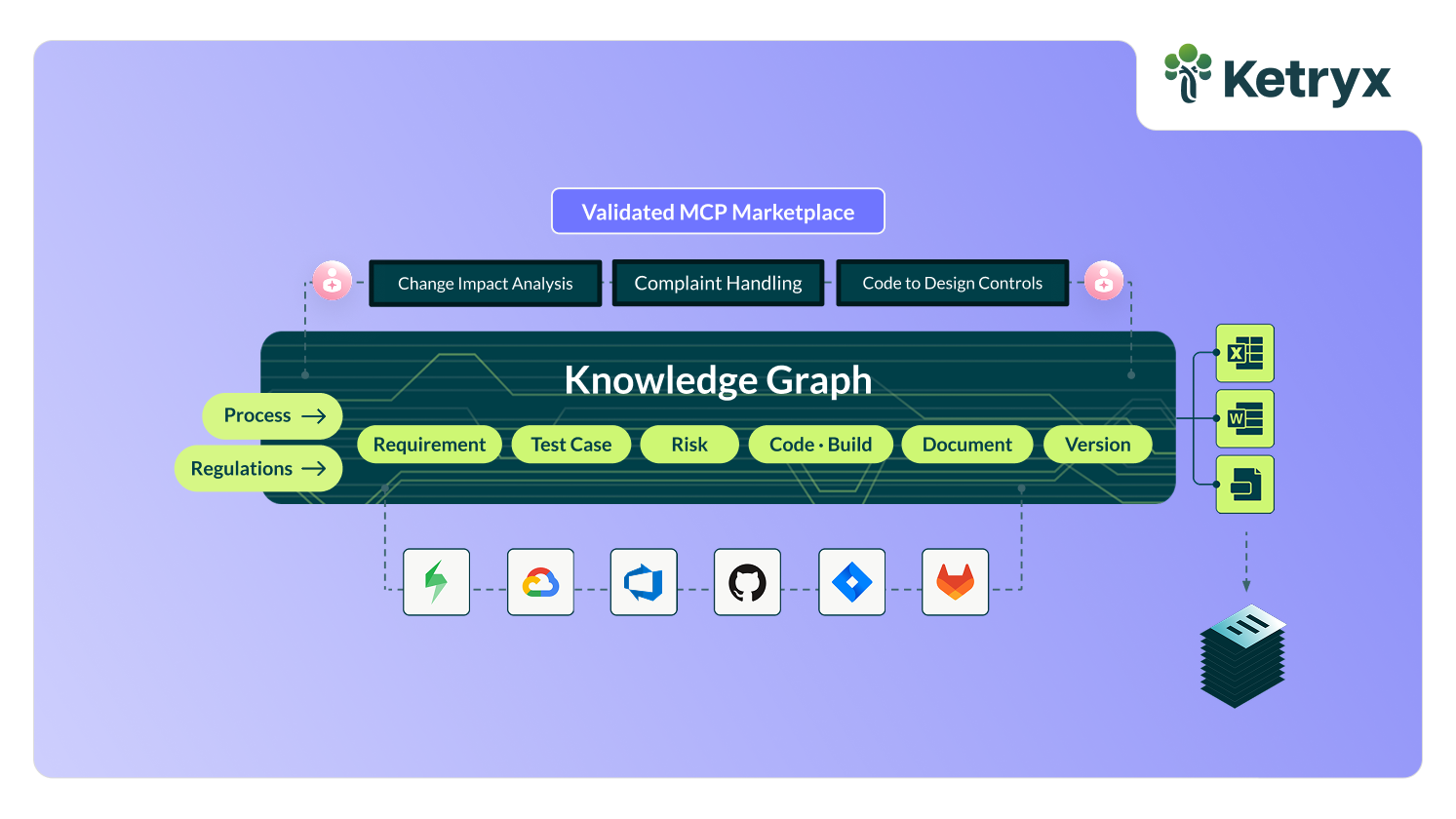
The AI custody chain and data integrity architecture
Picture the workflow most teams run today. A person opens an AI chat window and iterates: draft, edit, re-prompt, accept. Eventually, they copy the output into a document or ticket. A human working in a loop with AI is genuinely productive, and the FDA is not fundamentally objecting to that loop. They are responding to the chain of custody of AI-generated outputs.
The FDA issued several guidances on using AI in the product life cycle (link, link, link), which, along with existing regulatory requirements and standards, provide a good overview of the FDA’s expectations. Whether a record was generated with AI or not, it still needs to follow 21 CFR Part 11 (or Annex 11 in the EU), and be transitioned to a controlled record through a well-trained human review, an electronic signature tied to the reviewer’s identity, timestamp, version lock, and an immutable audit trail.
This is what data integrity means in practice. Part 11, EU Annex 11, and ALCOA++ all require controlled content to be owned by a qualified person. These regulatory frameworks mandate a custody chain, and AI-generated content is not an exception. The letter puts it plainly:
"If you use AI as an aid in document creation, you must review the AI-generated documents to ensure they were accurate and actually compliant with CGMP. Your failure to do so is a violation of 21 CFR 211.22(c). Overreliance on artificial intelligence for your drug manufacturing operations was also documented during the inspection."
That is not a restriction. It is a design spec that tells you exactly what you need to transform AI outputs into controlled data: a qualified, documented, and traceable approval step. The reviewer must know what “accurate” means for a given artifact, verify compliance with CGMP, and document the process.
Most companies have invested heavily in AI capabilities and rely on legacy processes and infrastructure to ensure this custody chain exists. The faster AI makes teams move, the harder it will be to utilize existing tools. The FDA is telling you how to approve properly. Build that into your process, and you can use AI as aggressively as you want.
We have been here before
Regulated life sciences experienced the same tension 15 years ago with cloud computing. Most quality leaders in the 2010s would say cloud-based infrastructure (the cloud) was incompatible with their compliance posture. Servers had to be on-premise, and you could not validate and audit a multi-tenant SaaS environment.
That consensus was wrong and caused costly mistakes and delays. Companies that avoided the cloud lost years of infrastructure modernization. The answer was never "don't use cloud." The answer was to establish necessary controls, validate your deployments, and document what is required.
We see a similar inflection point today with AI. The real question is about architecture: Part 11 electronic signatures on AI-drafted content, deterministic enforcement of approved SOPs, immutable audit trails from real development activity, and traceability that reflects the current state of the system rather than a point-in-time snapshot.
Teams that build that architecture in 2026 will have a two-year head start on everyone who waits for the regulatory picture to "stabilize." The waiting period is over, and the FDA just revealed what architecture looks like.
The neurosymbolic approach
I have been building and deploying AI for life sciences for more than a decade, most recently as Head of AI/ML at Amgen's medical device division before co-founding Ketryx. We started the company in 2021 on a specific bet: a foundation model alone would never be enough to accelerate regulated innovation.
End-to-end AI solutions offer immense power but have systematic limitations: simple mistakes, illogical responses, overreliance on historical training data, and a “black box” around intermediate decisions. Commercial LLMs can draft a traceability matrix that reads as if a senior V&V engineer wrote it, but they cannot tell you whether the output is verified and complete. You can’t move faster with AI that you don’t trust.
Ketryx closes that gap with a neurosymbolic architecture built for validation. A neural layer drafts while a symbolic layer enforces your QMS, standards, traceability relationships, and approval structure. This forces the model (Claude, ChatGPT, etc.) to stop acting like an uncontrolled content layer and instead act as a validated system that knows the rules, data, and requirements. Once a qualified human has approved the content, a deterministic algorithm generates downstream evidence with controlled data. It does so from human-approved outputs and enables teams to accelerate development without a compliance tax.
Three questions to bring back to your team
Every company I’ve talked to in the last 12 months is wondering how to introduce AI to accelerate their lifecycle. To better understand how to use AI and have it fit into your compliance workflows, I suggest asking your team these three questions:
- For any piece of AI-generated content that has entered one of our regulated records, can we produce the complete custody chain, from source data to human approval to the controlled state?
- Can we show, structurally, that no AI output can reach a controlled state without a qualified human review backed by a Part 11 electronic signature and an immutable audit trail?
- Are we taking a risk-based approach to where AI shows up in our lifecycle, with validation effort applied where the risk actually lives, and lighter-touch controls where it does not?
If any of those answers is uncertain, the work is architectural, not model selection.
Everything comes back to people
Why does any of this matter? It’s not because of the auditors overseeing your product. It’s for the people who use and depend on your product. Guardrails exist for the patient. Speed and safety don’t have to be a tradeoff when your architecture is right. They both serve the same goal: deliver safer products to patients faster than we did last year.
The warning letter is the FDA telling us publicly what that architecture has to include. Teams that read it as a spec are more empowered than ever to accelerate their development of safety-critical products.
How is your team handling the approval path for AI-generated content today? I'd like to hear where the architecture feels solid and where it feels like a gap.

Erez is passionate about improving patient care and health outcomes with software solutions. Over the last decade, Erez worked in industries including computational mathematics, biotech, and energy, helping build monitoring systems for pharmaceutical equipment and AI for medication management. Before Ketryx, Erez worked with Amgen, the world’s largest biotechnology company, as the head of AI/ML for their medical device division and with Wolfram Research, the builders of Mathematica and Wolfram|Alpha. Erez holds a Master of Science in Electrical Engineering and Computer Science and a Master of Business Administration from the Massachusetts Institute of Technology.